BRAKER 基因预测:BRAKER3、AUGUSTUS、GeneMark 和真核基因组注释
自动化真核基因组蛋白编码基因预测流程,整合 GeneMark、AUGUSTUS 以及转录组或蛋白证据。
30 秒判断
先看这四点,再决定要不要继续读完整评测。
BRAKER 不是单点工具,而是真核基因组注释流程里的基因预测环节。
最适合新物种基因组注释、蛋白编码基因预测和 RNA-seq/蛋白证据整合。
预测结果仍需 BUSCO、同源注释、基因长度分布和人工抽样检查。
先确认组装质量、重复序列屏蔽和证据数据,再运行 BRAKER3。
最适合已有真核基因组组装,并有 RNA-seq BAM、近缘物种蛋白或其他可用证据时,生成蛋白编码基因结构预测结果。
不适合细菌、古菌和多数病毒基因组注释;不适合没有完成组装质控、污染检查和证据质控就直接进入功能解释;也不适合人类临床变异报告、单细胞分析、影像 AI、宏基因组分类或直接诊断场景。
MAKER / AUGUSTUS / GeneMark
BRAKER 的搜索意图集中在 BRAKER3 和基因组注释流程
BRAKER 当前属于长尾页,优化重点不是夸功能,而是把它放进完整基因组注释流程:组装质量、重复序列屏蔽、RNA-seq/蛋白证据、基因预测、BUSCO 评估和功能注释。
- 流程位置:基因组组装和 repeat masking 之后,功能注释和比较基因组分析之前。
- 证据输入:RNA-seq、蛋白同源证据和物种相关训练数据会显著影响结果。
- 质控:用 BUSCO、基因长度分布、同源注释和人工抽样检查预测质量。
视频演示
适合谁用
适合从事真核新物种基因组注释、医学相关真菌/寄生虫/媒介生物研究、比较基因组和非模式生物组学分析的生信研究者、医学研究生、临床科研团队和 PI。
用它完成一次可复现数据分析
把分析过程留下来,而不只是导出一张漂亮图。
输入材料
一份清洗后的数据表和明确的统计问题
应该得到
分析代码/流程、结果表、图表和解释边界
- 1先写下变量定义、样本筛选和主要结局。
- 2选择合适的统计方法,并记录为什么这么选。
- 3生成结果表和图表,同时保存参数、版本和代码。
- 4把统计显著性、效应量和临床意义分开解释。
人工核验点
- 变量和样本数是否一致
- 方法是否符合数据类型
- 图表是否能被他人复现
更适合
最适合已有真核基因组组装,并有 RNA-seq BAM、近缘物种蛋白或其他可用证据时,生成蛋白编码基因结构预测结果。
不太适合
不适合细菌、古菌和多数病毒基因组注释;不适合没有完成组装质控、污染检查和证据质控就直接进入功能解释;也不适合人类临床变异报告、单细胞分析、影像 AI、宏基因组分类或直接诊断场景。
数据与隐私
BRAKER 通常在本地服务器或机构 HPC 上运行,基因组、RNA-seq 和蛋白证据不会因工具本身自动上传到第三方平台。若使用云服务器或共享集群,应按机构要求处理未发表基因组、病原体数据和可能涉及人源样本来源的测序数据,并记录数据存储、访问权限和计算环境。
医学科研场景
- 医学相关真核病原体基因组注释
- 真菌和寄生虫蛋白编码基因预测
- RNA-seq 证据辅助基因结构预测
- 媒介昆虫和非模式生物比较基因组
- 候选毒力、耐药和宿主互作基因筛选前的基因集构建
相关科研场景
查看全部场景核心功能
使用场景
优点与局限
优点
- +适合真核新物种从头注释:在没有成熟物种模型时,可根据目标基因组和证据数据训练或辅助预测参数。
- +能整合医学组学项目常见证据:RNA-seq BAM、蛋白同源证据和基因组序列可以进入同一基因预测流程。
- +输出结果便于衔接下游分析:GFF/GTF、蛋白 FASTA 和 CDS 文件可用于 BUSCO、功能注释、泛基因组和比较基因组分析。
- +自动化程度较高:相比手动训练 AUGUSTUS 或分别运行多个预测器,BRAKER 能减少重复配置和人工拼接步骤。
- +适合批量项目:对于多个近缘真核物种或多个菌株的统一注释,较容易形成可复现的命令行流程。
局限
- -安装和依赖配置有门槛:GeneMark、AUGUSTUS、ProtHint、SAMtools 等组件和许可证设置可能需要熟悉 Linux、生信环境管理和软件版本控制。
- -不适合原核基因组注释:细菌、古菌和多数病毒基因预测应优先考虑 Prokka、Bakta、PGAP 或病毒专用流程。
- -对输入质量敏感:低连续性组装、污染、未处理重复序列或质量较差的 RNA-seq 比对会影响基因边界和基因数量。
- -预测结果仍需验证:BRAKER 产生的是结构预测,不等于功能注释、致病机制解释或临床意义判断,需要结合 BUSCO、同源证据、表达证据和人工抽样检查。
- -不是网页式即开即用工具:医学科研团队如果缺少生信工程或 HPC 支持,可能需要较多时间处理安装、参数、队列脚本和报错排查。
快速上手
确认研究对象和问题:目标应为真核基因组,并明确 BRAKER 只用于蛋白编码基因结构预测,不用于临床诊断或人类变异解读。
检查输入数据:对 genome.fa 做组装连续性、污染、冗余、重复序列和 BUSCO 完整性评估;如使用 RNA-seq,先完成 reads 质控、比对并生成排序后的 BAM 文件。
准备运行环境:在 Linux 服务器或 HPC 上安装 BRAKER 及 GeneMark、AUGUSTUS、SAMtools、ProtHint 等依赖,确认环境变量、软件版本和 GeneMark 许可证可用。具体依赖名称、版本和参数应以官方 GitHub 文档为准。
选择证据模式并运行:根据项目数据选择基因组加 RNA-seq、基因组加蛋白证据或联合证据模式,为每个物种设置清晰的 species 名称、输入文件和输出目录。
质控并进入下游:检查 GFF/GTF、蛋白 FASTA 和 CDS 文件,用 BUSCO、基因数量、基因长度分布、同源注释比例和人工抽样判断结果是否可用于功能注释、比较基因组和论文分析。
详细介绍
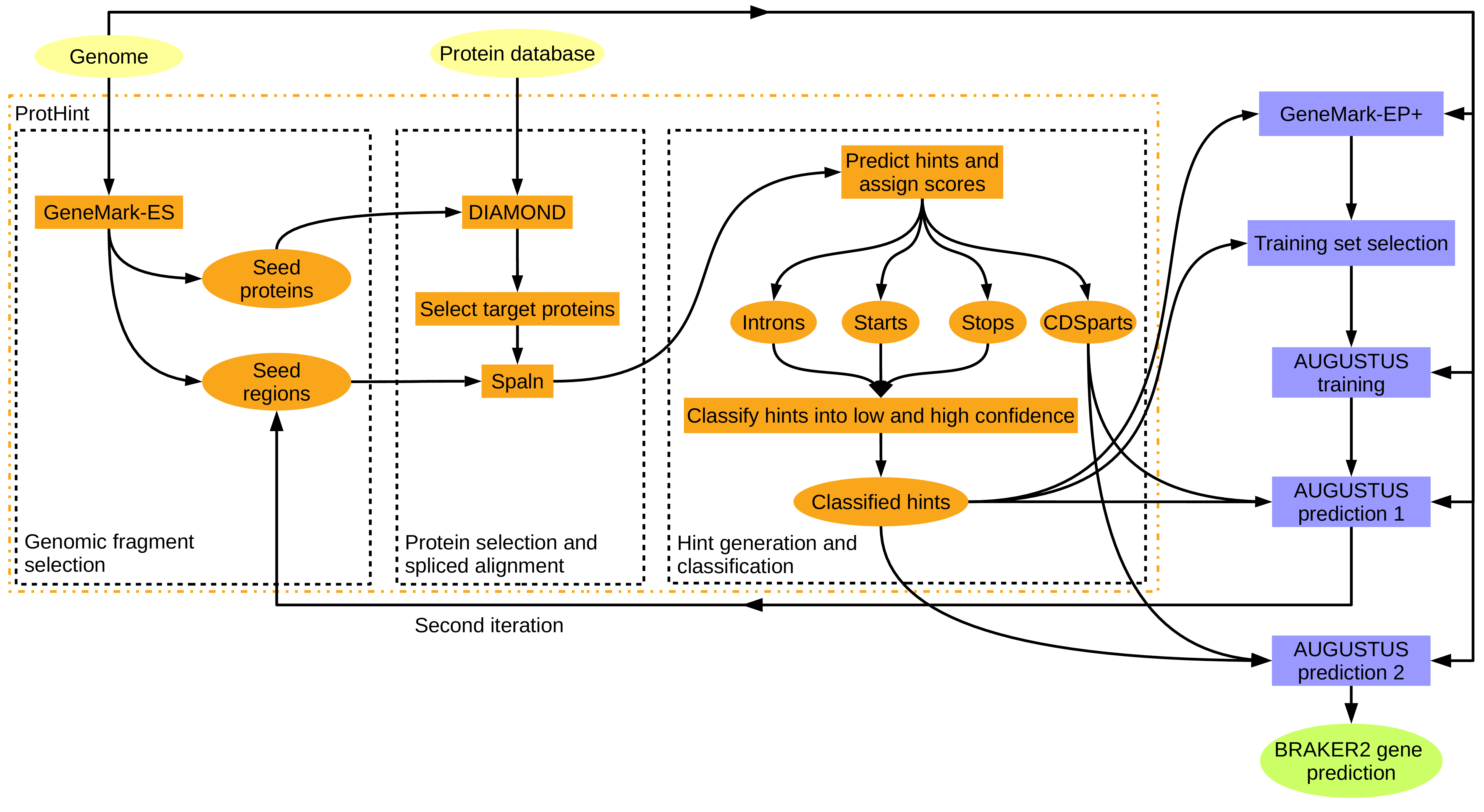
BRAKER 在基因组注释流程中的位置
BRAKER 是一个面向真核基因组的蛋白编码基因结构预测流程。它的核心任务是从基因组序列中识别可能的基因结构,包括外显子、内含子、编码序列和预测蛋白。
在医学科研中,BRAKER 主要服务于上游组学研究,而不是临床诊断。研究者通常在完成基因组组装、去污染、重复序列处理和基础质量评估后,再使用 BRAKER 生成初版结构注释。
BRAKER 常与 GeneMark、AUGUSTUS、RNA-seq 比对结果和蛋白同源证据配合使用。可以把它理解为结构注释流程中的自动化环节,用来减少手工训练、文件转换和多软件衔接的工作量。
需要强调的是,BRAKER 输出的是结构预测,不是功能注释。它可以提示哪些位置可能存在蛋白编码基因,但不能直接说明这些基因是否致病、是否耐药或是否可作为治疗靶点。
适合的医学科研场景
BRAKER 与临床医学的关系主要体现在病原体、媒介生物和非模式生物的基因组研究。对于新测序的医学相关真菌、寄生虫或节肢动物,往往没有高质量参考注释,此时 BRAKER 可以提供初版蛋白编码基因集。
例如,在机会致病真菌研究中,团队可能希望筛选转运蛋白、细胞壁相关蛋白、代谢酶或潜在毒力因子。BRAKER 生成的蛋白集合可以作为 InterProScan、eggNOG-mapper、BLAST 和 KEGG/GO 注释的输入。
在寄生虫或媒介昆虫研究中,BRAKER 可用于建立多个物种或多个株系的统一基因预测结果。这样有助于后续同源基因聚类、基因家族扩张、选择压力分析和候选抗原筛选。
对于医学相关非模式模型生物,例如感染模型、毒理模型或环境暴露研究对象,BRAKER 可帮助构建 GFF/GTF 和蛋白 FASTA,为 RNA-seq 定量、差异表达和功能富集提供参考基因集。
- 真核病原体研究:为真菌、寄生虫等物种建立蛋白编码基因集,用于耐药、毒力和宿主互作分析。
- 比较基因组:对多个近缘真核物种采用相近流程预测基因,减少不同注释来源导致的系统差异。
- 非模式生物组学:为缺乏参考注释的医学相关模型生物建立初版 GFF/GTF、CDS 和蛋白序列。
- 转录组辅助注释:利用 RNA-seq BAM 证据改善外显子边界和基因模型训练。
不适合的情况
BRAKER 不适合细菌、古菌和多数病毒基因组注释。原核生物基因结构与真核生物不同,通常应优先考虑 Prokka、Bakta、NCBI PGAP 或病毒专用注释流程。
它也不适合直接用于人类临床变异解读、肿瘤突变报告、药物基因组学报告或影像 AI 分析。BRAKER 的输出不能作为患者诊断、分型或用药建议的依据。
如果项目只有表达矩阵、单细胞转录组数据、宏基因组分类结果或已有人类参考基因组注释,BRAKER 通常不是合适入口。此类问题应选择差异表达、单细胞分析、宏基因组分类或临床变异注释工具。
对于组装质量较差、污染明显或重复序列未处理的基因组,直接运行 BRAKER 可能得到数量异常、结构破碎或假阳性较多的基因模型。此时应先回到组装和质控环节。
输入数据与证据选择
BRAKER 的基本输入通常包括真核基因组 FASTA 文件。根据项目条件,还可以加入 RNA-seq 比对生成的 BAM 文件,或来自近缘物种、公共数据库的蛋白序列证据。
RNA-seq 证据适合用于改善外显子、内含子和剪接边界,尤其是在研究对象缺乏成熟训练模型时更有价值。用于 BRAKER 前,RNA-seq reads 应完成质控、去接头、比对和排序索引。
蛋白证据适合没有本物种转录组数据,但存在近缘物种或同类群蛋白序列的项目。证据来源应尽量可信,避免混入远缘或污染来源序列,否则可能影响基因模型判断。
在医学病原体项目中,建议同时记录样本来源、测序平台、组装版本、证据数据版本和运行参数。后续论文审稿或重复分析时,这些信息往往比单次运行结果更重要。
结果质控与下游分析
BRAKER 常见输出包括 GFF/GTF 注释文件、预测蛋白 FASTA、CDS 序列和运行日志。研究者不应只看是否生成文件,而应检查基因数量、平均基因长度、内含子分布和异常短蛋白比例。
BUSCO 常用于评估基因集完整性,但 BUSCO 结果不能单独证明注释可靠。建议结合组装 BUSCO、蛋白同源注释比例、RNA-seq 支持情况和人工抽样查看,综合判断结果是否可用于论文。
对于候选毒力因子、耐药相关基因或宿主互作蛋白,BRAKER 只能提供候选蛋白序列。是否具有相关功能,需要进一步结合结构域、同源基因、表达变化、系统发育分析和实验验证。
如果研究涉及多个物种或多个菌株,最好使用一致的输入处理和参数策略。这样可以减少因为注释流程不同而导致的基因数量差异,从而提高比较基因组结论的可解释性。
安装、复现与数据合规
BRAKER 是命令行工具,对 Linux、软件依赖、环境变量和文件路径管理有要求。GeneMark、AUGUSTUS、ProtHint、SAMtools 等依赖的安装方式和版本要求应以官方 GitHub 文档为准。
对于医学研究生和临床团队,建议在机构 HPC、实验室服务器或可审计的云环境中运行,并把命令、软件版本、输入文件校验值和输出目录结构写入分析记录。
BRAKER 通常在本地或集群运行,工具本身不会自动上传基因组或 RNA-seq 数据。若样本来自患者相关材料、未发表病原体或敏感地区采样,应遵守伦理审批、数据使用协议和机构安全要求。
实用判断:如果你的问题是“这个真核非模式生物基因组里有哪些蛋白编码基因”,BRAKER 可能合适;如果问题是“这个患者突变是否致病”或“这张影像是否提示疾病”,BRAKER 不是合适工具。
与常见替代工具的区别
MAKER 更偏完整注释框架,适合需要多轮整合从头预测、转录本、蛋白同源和人工修订的项目。它的灵活性较高,但流程设计和维护成本也可能更高。
AUGUSTUS 和 GeneMark 可以独立用于基因预测,也常作为 BRAKER 背后的关键组件。对于希望精细控制训练模型和参数的生信研究者,直接使用这些工具可能更灵活。
Funannotate 在真菌基因组项目中较常见,覆盖结构注释、功能注释和部分质控步骤。若研究对象主要是真菌,并希望流程覆盖更多下游注释环节,可以把它作为对照方案评估。
| 工具 | 更适合的问题 |
| BRAKER | 真核基因组蛋白编码基因结构预测,尤其是有 RNA-seq 或蛋白证据时。 |
| MAKER | 需要多证据整合、迭代和人工修订的较完整注释项目。 |
| Funannotate | 真菌基因组从结构注释到功能注释的流程化分析。 |
替代选择
如果 BRAKER 不适合你,可以考虑:
同类工具推荐
如果你需要更完整的文献工作流
从检索到精读,一站完成
这个工具适合特定场景。如果你需要中文检索、实时翻译、AI 辅助精读,可以试试超能文献。
了解超能文献